
How to Harmonize Multi-Omics Data Without Losing Your Mind
Why Multi-Omics Data Harmonization Feels Impossible — And How to Fix It
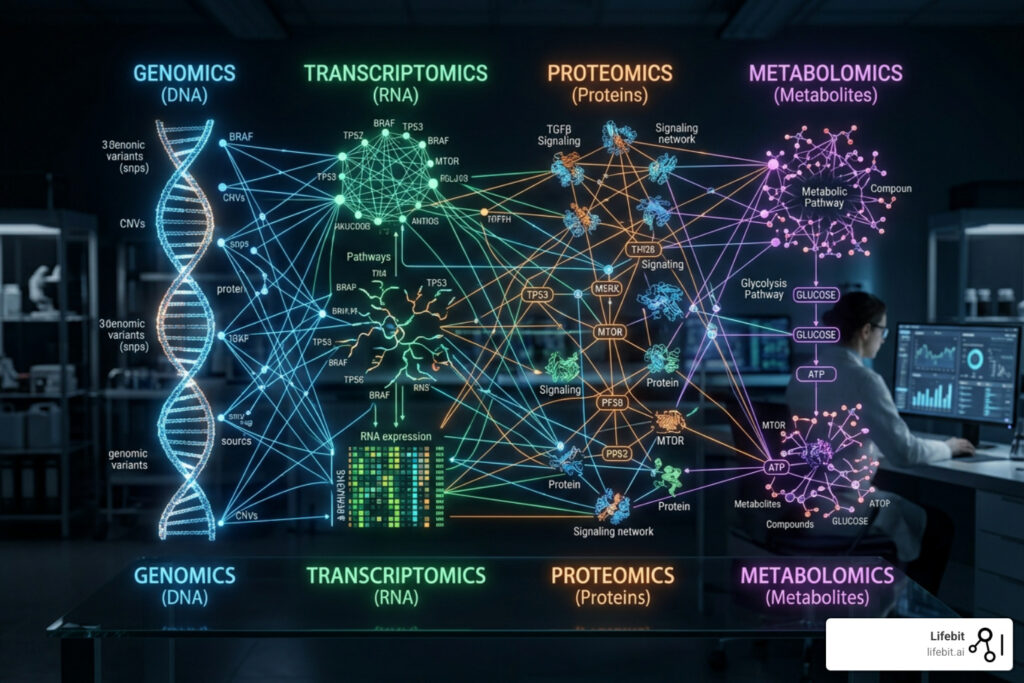
Data harmonization multi-omics is the process of standardizing, normalizing, and integrating biological datasets from multiple “omic” layers — such as genomics, transcriptomics, proteomics, and metabolomics — so they can be analyzed together in a meaningful way.
Here’s a quick overview of what it involves:
| Step | What It Means |
|---|---|
| Standardize formats | Convert raw data from different platforms into a common structure |
| Remove batch effects | Correct for technical noise introduced by different labs or instruments |
| Handle missing values | Fill gaps or filter incomplete data without biasing results |
| Normalize distributions | Align data scales across different omic layers |
| Integrate datasets | Combine layers to reveal biological signals no single dataset could show alone |
Biological data is messy. A genomics file from one lab rarely looks like a proteomics file from another. Add in metabolomics, epigenomics, and clinical records — each with different formats, platforms, and quality standards — and you have a data integration nightmare.
This isn’t a minor inconvenience. Poorly harmonized multi-omics data produces unreliable results. It slows down drug discovery, muddies biomarker signals, and can derail entire research programs before they get off the ground.
For pharma teams, public health institutions, and regulatory bodies, the stakes are especially high. You’re not just managing messy spreadsheets — you’re working with sensitive, siloed datasets across institutions, jurisdictions, and compliance frameworks. Getting harmonization wrong doesn’t just waste time. It wastes resources, delays treatments, and erodes trust in the data itself.
I’m Dr. Maria Chatzou Dunford, CEO and Co-founder of Lifebit, and with over 15 years in computational biology and bioinformatics — including core contributions to Nextflow and biomedical data integration research at the Centre for Genomic Regulation — data harmonization multi-omics is a challenge I’ve spent my career building solutions for. In this guide, I’ll walk you through exactly how to do it right.

Simple data harmonization multi-omics glossary:
Why Multi-Omics Data Harmonization is the Backbone of Modern Research
In the era of modern bioinformatics, looking at a single layer of biology is like trying to understand a complex engine by looking at only the spark plugs. While genomics tells us what might happen (the blueprint), transcriptomics, proteomics, and metabolomics tell us what is happening (the execution and the outcome). The real magic happens at the intersection of these layers, where the flow of biological information from DNA to RNA to protein to metabolite can be traced in real-time.
Precision medicine relies on this holistic view. By integrating diverse datasets, we can move toward a systems biology approach that identifies the true drivers of disease rather than just the symptoms. This is essential for biomarker discovery; a single genetic variant might be interesting, but a variant that correlates with a specific protein expression and a metabolic shift is a potential drug target with a much higher probability of clinical success. This approach is often referred to as “P4 Medicine”—Predictive, Preventive, Personalized, and Participatory—and it is entirely dependent on the quality of the underlying data integration.
However, the road to these insights is paved with technical hurdles. Researchers must weigh the advantages and tradeoffs of multi-omics, balancing the richness of the data against the sheer complexity of integrating it. Without proper data harmonization multi-omics, we risk finding “signals” that are actually just artifacts of how the data was collected. For instance, a perceived difference in gene expression between two patient cohorts might actually be a result of different sequencing depths or library preparation kits rather than the underlying pathology. Harmonization acts as the filter that separates this technical noise from the biological truth, ensuring that the conclusions drawn by researchers are both reproducible and clinically relevant. Furthermore, as we move toward including the microbiome and epigenome into these models, the dimensionality of the data increases exponentially, making the need for robust, automated harmonization even more critical.
Overcoming the 3 Biggest Roadblocks in Data Integration
When we set out to integrate multi-omic data, we usually hit three massive walls. Understanding these is the first step toward overcoming them. These roadblocks are not just technical; they are fundamental challenges in how biological information is captured and stored across the global research community.
Batch Effects: This is the “silent killer” of biological research. Batch effects occur when non-biological factors—like the day of the week a sample was processed, the specific technician involved, the reagent lot used, or even the ambient temperature of the lab—create artificial variations in the data. In large-scale studies, these can easily swamp the actual biological signal. Mitigating batch effects is perhaps the most critical task in the entire harmonization pipeline. If you combine two datasets without correcting for batch effects, your machine learning model will likely learn to distinguish between the two labs that produced the data rather than the two diseases you are trying to study.
Incompatible Data Formats and Metadata: Genomics uses FASTQ and VCF; proteomics might use mzML or simple CSVs. Beyond the file extensions, the metadata—the “data about the data”—is often inconsistent. One dataset might label a patient’s age in years, while another uses birth dates. One might use the term “neoplasm” while another uses “tumor.” Standardizing these into a unified ontology (like SNOMED CT or the Human Phenotype Ontology) is manual, grueling work if you don’t have the right tools. This lack of semantic interoperability is a major bottleneck in cross-institutional collaborations.
Missing Values and Experimental Design: Not every patient in a study will have every type of “omic” data. You might have genomics for 1,000 people but proteomics for only 200. This is often referred to as the “missingness” problem. In proteomics, missing values are often “not at random” (MNAR), meaning a protein might be missing from the data simply because its concentration was below the detection limit of the mass spectrometer. Handling these gaps without introducing bias requires sophisticated statistical approaches, such as multiple imputation by chained equations (MICE) or K-nearest neighbors (KNN) imputation, tailored to the specific omic layer.
Solving the Batch Effect Crisis with TAMPOR and ComBat
To tackle the batch effect issue, two main contenders often come up: TAMPOR and ComBat. Both aim to remove technical variation while preserving biological variation, but they do so using different mathematical philosophies.
TAMPOR (Targeted Analysis of Multi-Platform Omics Data) is an algorithm designed to handle the nuances of protein measurements across different platforms. It focuses on median variance and uses robust statistical methods to ensure that the adjustments don’t accidentally “smooth over” the real biology. It is particularly effective when dealing with large-scale proteomics where the distribution of data might not be perfectly normal.
On the other hand, ComBat (and its newer iteration, ComBat-seq for RNA-seq data) uses Empirical Bayes inference to estimate and remove batch effects. It is highly effective when sample sizes are small, as it “borrows” information across genes to make more stable estimates of the batch parameters.
| Feature | TAMPOR | ComBat |
|---|---|---|
| Statistical Core | Median-based variance adjustment | Empirical Bayes |
| Primary Use Case | Multi-platform proteomics | General gene expression/transcriptomics |
| Resilience | High resilience to outliers | Sensitive to small sample sizes |
| Assumption | Data follows a robust median distribution | Data follows a normal distribution |
| Handling of Covariates | Can preserve known biological groups | Excellent at protecting known biological variation |
Step-by-Step Best Practices for Effective Data Harmonization Multi-Omics
Achieving high-quality data harmonization multi-omics isn’t about one single “magic” tool; it’s about a disciplined pipeline. We follow a rigorous process to ensure that when we look at integrated data, we are seeing nature, not noise. This pipeline must be reproducible, meaning that another researcher should be able to take your raw data and harmonization script and arrive at the exact same result.
Standardization and Coordinate Alignment: Before anything else, move all data into a common coordinate system. For genomics, this means ensuring all datasets are mapped to the same genome assembly (e.g., GRCh38). For proteomics and metabolomics, it means using standardized vocabularies (like Gene Ontology terms or ChEBI IDs) to ensure that “Protein A” in one study is the same as “Protein A” in another.
Data Quality Control (QC): Run rigorous checks at both the sample and feature levels. If a sample has a low read depth in genomics or too many missing peaks in proteomics, it might need to be discarded. We also look for “outlier” samples that don’t fit the expected distribution, which could indicate contamination or sample swaps. QC is not a one-time step; it happens after every major transformation of the data.
Normalization and Scaling: This step ensures that measurements are comparable across different scales. For instance, transcriptomics data might be measured in counts per million (CPM), while proteomics is measured in intensity values. You might use log-transformation to stabilize variance or Z-score scaling to ensure that a high-abundance protein doesn’t mathematically overshadow a low-abundance but biologically critical metabolite. Methods like Quantile Normalization are often used to make the distributions of different samples identical.
Compatibility Mapping and ID Resolution: Ensure that your identifiers match across layers. Mapping a protein ID (Uniprot) to a gene ID (Ensembl) and then to a transcript ID is a classic “bioinformatics headache.” This requires up-to-date mapping databases and an understanding of isoforms—where one gene can produce multiple different proteins with different functions.

Selecting High-Quality Datasets for Seamless Data Harmonization Multi-Omics
You can’t harmonize garbage into gold. The success of your project depends heavily on the quality of your source data. Fortunately, the scientific community has built incredible repositories that serve as the gold standard for multi-omics research. These repositories often have their own internal harmonization standards, which makes your job significantly easier:
- The Cancer Genome Atlas (TCGA): A landmark program that has generated over 2.5 petabytes of genomic, epigenomic, transcriptomic, and proteomic data across 33 different cancer types. It is the most comprehensive multi-omics resource available today.
- International Cancer Genome Consortium (ICGC): Focused on defining the genomes of 50 different cancer types of clinical and societal importance across the globe.
- Gene Expression Omnibus (GEO) and ArrayExpress: These are the primary archives for functional genomics data. While they contain a wealth of data, they require significant harmonization because the data is submitted by thousands of different labs using different protocols.
- CPTAC Portal: The Clinical Proteomic Tumor Analysis Consortium is the go-to for high-quality proteomic data. What makes CPTAC special is that its samples are often the same ones used in TCGA, allowing for direct proteogenomic integration.
- UK Biobank: A massive longitudinal study containing deep genetic and health information on 500,000 participants, increasingly including proteomics and metabolomics data.
Top Tools and Software to Automate Your Harmonization Workflow
Manually coding a harmonization pipeline for every project is a recipe for burnout and error. We recommend leveraging established software packages that have been battle-tested by the community. These tools are designed to handle the high dimensionality and sparsity of multi-omics data.
- mixOmics: This R package is a powerhouse for feature selection and data integration. It offers a suite of multivariate methods, such as PLS (Partial Least Squares) and CCA (Canonical Correlation Analysis), which are particularly good at finding correlations between different omic layers. It allows researchers to identify a small subset of variables that explain the most variation across datasets.
- Harmony and Scanorama: If you are working with single-cell data, these tools are indispensable. Single-cell experiments are notoriously prone to batch effects. Harmony uses an iterative clustering approach to align cells from different experiments into a shared space, while Scanorama uses techniques inspired by computer vision to “stitch” together disparate datasets.
- MOFA+ (Multi-Omics Factor Analysis): This is a framework for the unsupervised integration of multi-omics data sets. MOFA+ allows you to discover the principal sources of variation (factors) across the different layers. For example, it can identify a factor that represents “inflammation” which is visible in both the transcriptome and the proteome, while also identifying factors that are specific to only one layer.
Future Trends in Data Harmonization Multi-Omics and AI
The future of data harmonization multi-omics is being written by Artificial Intelligence. We are moving away from simple linear corrections and toward deep generative models that can learn the complex, non-linear relationships between biological layers.
One exciting area is graph-linked embedding, which represents biological entities (genes, proteins, drugs) as nodes in a massive network. This allows AI to understand the context of a data point—for example, knowing that a specific protein is part of a certain signaling pathway—rather than just treating it as a numerical value in a spreadsheet.
Tools like MultiVI (a Variational Autoencoder) are now being used for single-cell multi-omics integration, allowing researchers to look at chromatin accessibility and gene expression simultaneously. This level of detail is helping us understand cell-type-specific impacts of splicing aberrations in diseases like clonal hematopoiesis. These aberrations were previously invisible to bulk sequencing methods, which average out the signals from millions of cells, potentially masking the rare cell populations that drive disease progression.
Real-World Impact: From Cancer Subtyping to COVID-19 Targets
Why do we go through all this trouble? Because when data harmonization multi-omics is done correctly, it saves lives and accelerates the delivery of new therapies to patients who need them most.
In oncology, researchers use tools like Nemo for cancer subtyping by integration of partial data. Often, clinical trials have missing data for some patients—perhaps a tissue sample was too small for proteomics but large enough for genomics. Nemo allows researchers to group patients into subtypes even when their data is incomplete. This has led to the discovery of new breast cancer subtypes that respond differently to chemotherapy, allowing doctors to tailor treatments more effectively.
During the COVID-19 pandemic, multi-omics harmonization allowed scientists to rapidly identify key inflammatory markers and potential drug-induced liver injury risks. By integrating viral genomics with host transcriptomics and proteomics, researchers could pinpoint exactly how the virus hijacked cellular machinery and which patients were at the highest risk for “cytokine storms.” This data was instrumental in repurposing existing drugs like dexamethasone for severe cases.
In neurodegenerative research, harmonizing multi-platform proteomics has been vital for studying Alzheimer’s Disease (AD). By looking at AD-associated proteins like APP and MAPT MTBR across different cohorts from around the world, scientists are finally getting a clearer picture of the protein aggregation pathways that lead to cognitive decline. Harmonization allows us to see that these pathways are remarkably consistent across different populations, suggesting that a single therapeutic approach might work for a broad range of patients. Furthermore, in the realm of rare diseases, harmonization allows researchers to combine data from tiny patient populations across multiple countries, providing the statistical power needed to identify the genetic cause of a disease that might only affect a few dozen people globally.
Frequently Asked Questions about Multi-Omics Harmonization
What are the main limitations of current harmonization methods?
The biggest limitation is the “black box” nature of some AI and deep learning methods. While they are incredibly powerful at aligning data, it can be difficult for a biologist to understand exactly how the data was transformed. This lack of interpretability can be a hurdle for regulatory approval. There is also the “n < p" problem, where we have many more features (tens of thousands of genes/proteins) than samples (often only a few hundred patients), which can lead to overfitting if not handled with care.
How does harmonization accelerate pharmaceutical research?
By providing a “clean,” integrated dataset, harmonization allows pharma teams to run virtual cross-trial analyses. This means they can compare the results of a new drug candidate against historical data from dozens of other studies. This “external control arm” approach can significantly speed up the identification of safety signals or efficacy markers, potentially reducing the need for large, expensive Phase III trials.
Can TAMPOR handle multi-platform proteomics?
Yes, TAMPOR was specifically designed to handle the challenges of multi-platform proteomics. It is excellent at aligning protein abundance measurements across different mass spectrometry runs or even different technological platforms (like Olink’s proximity extension assay vs. SomaScan’s aptamer-based technology). It ensures that the biological signal remains consistent regardless of the measurement tool.
Is cloud computing necessary for multi-omics harmonization?
While not strictly necessary for small datasets, cloud computing is becoming essential for large-scale multi-omics. The sheer volume of data (often petabytes) and the computational intensity of algorithms like MOFA+ or deep learning models require the scalable resources that only the cloud can provide. Moreover, cloud environments facilitate the secure sharing of data between international collaborators.
How do you handle data privacy during harmonization?
Data privacy is handled through a combination of de-identification, encryption, and federated analysis. Instead of moving sensitive patient data to a central server, federated AI allows the model to “travel” to the data. The harmonization happens locally within a secure environment, and only the non-sensitive, aggregated results are shared. This ensures compliance with strict regulations like GDPR and HIPAA.
Conclusion: Future-Proofing Your Research with Federated AI
The volume of biological data is growing exponentially, but our ability to move it is hitting a physical limit. The future of data harmonization multi-omics isn’t about moving all the data to one central “bucket”—it’s about federated AI.
At Lifebit, we believe that the data should stay where it is, especially when dealing with sensitive patient information in the UK, USA, Europe, or Singapore. Our platform provides a Trusted Research Environment (TRE) and a Trusted Data Lakehouse (TDL) that allows researchers to bring their analysis to the data.
With our R.E.A.L. (Real-time Evidence & Analytics Layer), we enable secure, federated governance and real-time insights across hybrid data ecosystems. This means you can harmonize and analyze data across five continents without the security risks or costs of data egress.
By adopting a federated approach, you aren’t just solving today’s harmonization headache; you are future-proofing your research for a world where data is global, decentralized, and more powerful than ever.
Ready to stop wrestling with your data and start discovering? Explore how Lifebit can transform your multi-omics workflow.